4.4. Exercise: Run a local BLAST+ database search#
This is an exercise in using the command-line to accomplish a task. You will be making use of the commands discussed earlier.
Task
Given a protein sequence, identify matching sequences from a protein sequence database.
Approach
Using programs in the NCBI BLAST+ package, you can search a database of sequences using sequence (query) to identify matching sequences.
4.4.1. Summary of steps#
Install NCBI BLAST+
Download protein query sequence
Download protein sequence database and format it
Search database using the query sequence
4.4.2. Get sample data#
To proceed, you will need to download the protein query sequence and database used in this exercise.
Download query sequence#
The protein query sequence used in this exercise is Spike glycoprotein from Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). It is available from UniProtKB — the protein knowledge base.
The database identifier for this protein is P0DTC2. You can download the sequence in FASTA format from the entry page or using this direct link:
Download protein database#
The database used in this exercise is UniProtKB Swiss-Prot. It is a manually annotated database of protein sequences with added functional information.
You can download the entire database as a compressed FASTA format file from the downloads page on the website.
4.4.3. Install the NCBI BLAST+ program#
Attention
This procedure installs software in system paths and so requires administrator privileges.
NCBI BLAST+ is available in the Linux package
repositories. You can install it using apt:
sudo apt install ncbi-blast+
Type y when prompted to continue.
[sudo] password for user:
Reading package lists... Done
Building dependency tree
Reading state information... Done
The following additional packages will be installed:
libmbedcrypto3 libmbedtls12 libmbedx509-0 ncbi-data
The following NEW packages will be installed:
libmbedcrypto3 libmbedtls12 libmbedx509-0 ncbi-blast+ ncbi-data
0 upgraded, 5 newly installed, 0 to remove and 0 not upgraded.
Need to get 14.9 MB of archives.
After this operation, 75.0 MB of additional disk space will be used.
Do you want to continue? [Y/n] y
4.4.4. Download query sequence#
You can follow these steps to download the query sequence:
Create new directory
Change into it
Download query sequence
View the downloaded sequence (optional)
Create new directory: mkdir#
To keep the input and output files related to this project together, create a new directory in your home directory using the mkdir command.
mkdir -p ~/projects/sars-cov-2
Here:
~ is shortcut for home directory.
-p creates parent directories if necessary.
In this case, the projects directory does not
exist, so it is also created.
Change directory: cd#
Change into the newly created directory using the cd command:
cd ~/projects/sars-cov-2
Download sequence: wget#
To download the sequence file, you can use the
wget command with the link to download as the
argument. In this case, the link to download is the URL
corresponding to the FASTA format file
(see sample data):
wget https://www.uniprot.org/uniprot/P0DTC2.fasta
When the download is complete, you can use the ls command to verify if the file exists:
ls -l
Output:
total 4
-rw-rw-r-- 1 user user 1414 Feb 10 00:00 P0DTC2.fasta
View downloaded sequence: cat or less#
Since P0DTC2.fasta is in FASTA format — a plain-text
format, you can use the
cat command to view the
file’s contents:
cat P0DTC2.fasta
Output:
>sp|P0DTC2|SPIKE_SARS2 Spike glycoprotein OS=Severe acute respiratory syndrome coronavirus 2 OX=2697049 GN=S PE=1 SV=1
MFVFLVLLPLVSSQCVNLTTRTQLPPAYTNSFTRGVYYPDKVFRSSVLHSTQDLFLPFFS
NVTWFHAIHVSGTNGTKRFDNPVLPFNDGVYFASTEKSNIIRGWIFGTTLDSKTQSLLIV
NNATNVVIKVCEFQFCNDPFLGVYYHKNNKSWMESEFRVYSSANNCTFEYVSQPFLMDLE
GKQGNFKNLREFVFKNIDGYFKIYSKHTPINLVRDLPQGFSALEPLVDLPIGINITRFQT
LLALHRSYLTPGDSSSGWTAGAAAYYVGYLQPRTFLLKYNENGTITDAVDCALDPLSETK
CTLKSFTVEKGIYQTSNFRVQPTESIVRFPNITNLCPFGEVFNATRFASVYAWNRKRISN
CVADYSVLYNSASFSTFKCYGVSPTKLNDLCFTNVYADSFVIRGDEVRQIAPGQTGKIAD
YNYKLPDDFTGCVIAWNSNNLDSKVGGNYNYLYRLFRKSNLKPFERDISTEIYQAGSTPC
NGVEGFNCYFPLQSYGFQPTNGVGYQPYRVVVLSFELLHAPATVCGPKKSTNLVKNKCVN
FNFNGLTGTGVLTESNKKFLPFQQFGRDIADTTDAVRDPQTLEILDITPCSFGGVSVITP
GTNTSNQVAVLYQDVNCTEVPVAIHADQLTPTWRVYSTGSNVFQTRAGCLIGAEHVNNSY
ECDIPIGAGICASYQTQTNSPRRARSVASQSIIAYTMSLGAENSVAYSNNSIAIPTNFTI
SVTTEILPVSMTKTSVDCTMYICGDSTECSNLLLQYGSFCTQLNRALTGIAVEQDKNTQE
VFAQVKQIYKTPPIKDFGGFNFSQILPDPSKPSKRSFIEDLLFNKVTLADAGFIKQYGDC
LGDIAARDLICAQKFNGLTVLPPLLTDEMIAQYTSALLAGTITSGWTFGAGAALQIPFAM
QMAYRFNGIGVTQNVLYENQKLIANQFNSAIGKIQDSLSSTASALGKLQDVVNQNAQALN
TLVKQLSSNFGAISSVLNDILSRLDKVEAEVQIDRLITGRLQSLQTYVTQQLIRAAEIRA
SANLAATKMSECVLGQSKRVDFCGKGYHLMSFPQSAPHGVVFLHVTYVPAQEKNFTTAPA
ICHDGKAHFPREGVFVSNGTHWFVTQRNFYEPQIITTDNTFVSGNCDVVIGIVNNTVYDP
LQPELDSFKEELDKYFKNHTSPDVDLGDISGINASVVNIQKEIDRLNEVAKNLNESLIDL
QELGKYEQYIKWPWYIWLGFIAGLIAIVMVTIMLCCMTSCCSCLKGCCSCGSCCKFDEDD
SEPVLKGVKLHYT
For more control, you can use the
less command instead of cat.
4.4.5. Download protein sequence database#
You can follow these steps to download and prepare the protein sequence database:
Create new directory
Change into it
Download the database archive
Uncompress (or extract) the database archive
Format the database
Create new directory: mkdir#
In order to keep all BLAST databases in one location, create a directory to store them using the mkdir command:
mkdir ~/databases
Change directory: cd#
Change into the newly created directory using the cd command:
cd ~/databases
Download the database archive: wget#
Visit the database downloads page on the UniProt website.
Navigate to the UniProtKB section.
Right-click on the fasta download link corresponding to Reviewed (Swiss-Prot) and then copy it to clipboard (Fig. 83).
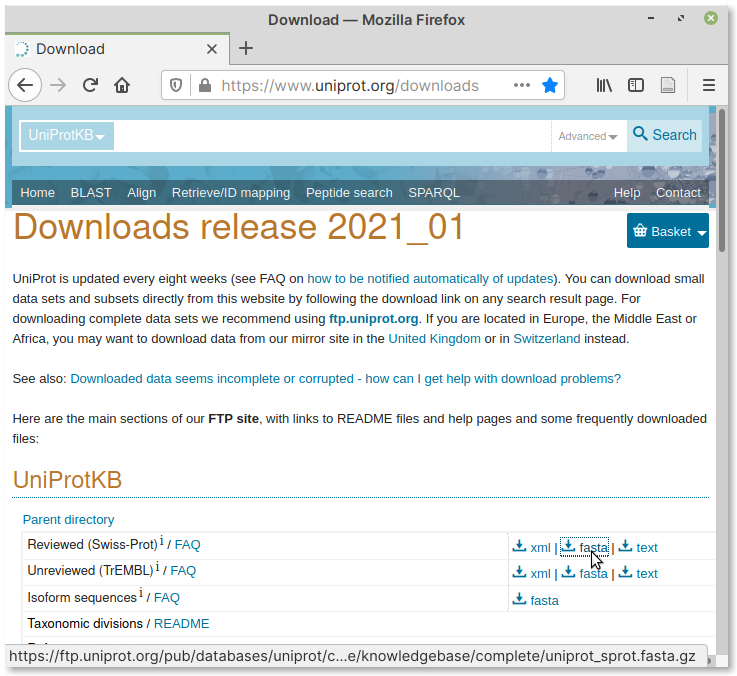
Fig. 83 Download link for Swiss-Prot database#
To download the database, you can use the
wget command with the link to download as the
argument:
wget https://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/complete/uniprot_sprot.fasta.gz
When the download is complete, you will find a file
named uniprot_sprot.fasta.gz in the current directory.
You can use the ls command to verify
if it exists:
ls -lh
Output:
total 86M
-rw-rw-r-- 1 user user 86M Feb 10 15:00 uniprot_sprot.fasta.gz
Since this file is in a compressed format (.gz),
you will need to uncompress it before proceeding.
Uncompress the database archive: gunzip#
To uncompress (or extract) the database archive file
downloaded in the previous step, you can use the
gunzip command.
Note
By default, gunzip will remove the original compressed file after extraction.
If you would like to keep the original file (.gz),
you can include the -k (keep input files) option
with gunzip.
Provide the file name of the downloaded file as the argument:
gunzip uniprot_sprot.fasta.gz
When the extraction is complete, you will find the database file in FASTA format in the same directory:
ls -lh
Output:
total 267M
-rw-rw-r-- 1 user user 267M Feb 10 15:00 uniprot_sprot.fasta
View the database#
Since this extracted database file is large, you can use the head command to view the first few lines of the file:
head -n 5 uniprot_sprot.fasta
Output:
>sp|Q6GZX4|001R_FRG3G Putative transcription factor 001R OS=Frog virus 3 (isolate Goorha) OX=654924 GN=FV3-001R PE=4 SV=1
MAFSAEDVLKEYDRRRRMEALLLSLYYPNDRKLLDYKEWSPPRVQVECPKAPVEWNNPPS
EKGLIVGHFSGIKYKGEKAQASEVDVNKMCCWVSKFKDAMRRYQGIQTCKIPGKVLSDLD
AKIKAYNLTVEGVEGFVRYSRVTKQHVAAFLKELRHSKQYENVNLIHYILTDKRVDIQHL
EKDLVKDFKALVESAHRMRQGHMINVKYILYQLLKKHGHGPDGPDILTVKTGSKGVLYDD
Alternatively, you can use the less command to view it one page at a time:
less uniprot_sprot.fasta
If you would like to count the number of sequences in the
database, you can use the grep command.
grep ">" -c uniprot_sprot.fasta
Output:
564277
The -c option of grep, counts the number of
times the given search string (> in this case)
occurs in the input file.
Note
A sequence in a FASTA format should start with
the > character. Hence, counting the number of times
it occurs gives the number of sequences in the file.
You can now proceed towards formatting the database.
Format database for BLAST search: makeblastdb#
The database needs to be formatted before it can be
used in a BLAST search. You can format it using the
makeblastdb command, which is part of the
NCBI BLAST+ package.
The command has multiple options. Here is an example:
makeblastdb -in uniprot_sprot.fasta -parse_seqids \
-title "Swiss-Prot" -dbtype prot -out swissprot
Note
The \ character splits the long command into
multiple lines.
Output:
Building a new DB, current time: 03/24/2021 15:12:50
New DB name: /home/user/databases/swissprot
New DB title: Swiss-Prot
Sequence type: Protein
Keep MBits: T
Maximum file size: 1000000000B
Adding sequences from FASTA; added 564277 sequences in 47.507 seconds.
What the options mean:
-inFile name containing input sequences.
-parse_seqidsParse sequence identifiers from the input file. These will be displayed in search results.
-titleA descriptive name for this database.
-dbtypeThe type of input sequences — acceptable values are
prot(for protein) andnucl(for nucleotide) sequences.-outThe value here will be used to name the output files. This is also the name you will need to use for the database while doing a search (see New DB Name) in output.
When formatting is complete, you will notice the following
files in the databases directory:
ls -lh
Output:
total 585M
-rw-rw-r-- 1 user user 100M Mar 24 15:13 swissprot.phr
-rw-rw-r-- 1 user user 4.4M Mar 24 15:13 swissprot.pin
-rw-rw-r-- 1 user user 2.2M Mar 24 15:13 swissprot.pog
-rw-rw-r-- 1 user user 18M Mar 24 15:13 swissprot.psd
-rw-rw-r-- 1 user user 411K Mar 24 15:13 swissprot.psi
-rw-rw-r-- 1 user user 195M Mar 24 15:13 swissprot.psq
-rw-rw-r-- 1 user user 267M Feb 10 15:00 uniprot_sprot.fasta
4.4.6. Search database using query sequence#
With the query sequence downloaded and the database downloaded and formatted, you can start performing a BLAST search.
First, change into the directory containing the query sequence:
cd ~/projects/sars-cov-2
Now run the blastp command using the query sequence
and the complete path to the database:
blastp -query P0DTC2.fasta \
-db /home/user/databases/swissprot \
-out blastp-results.txt \
-outfmt "7 sacc stitle qlen slen pident"
What the options mean:
-queryPath to the query sequence.
-dbComplete path of the sequence database.
-outFile to save results to.
-outfmtFormat of the output file. This will use format option
7(tab-delimited text) and include the following information:accession number and description of matching sequences (
saccandstitle),query and subject sequence lengths (
qlenandslen)percentage identity of the match (
pident).
When the database search is complete, you can open
blastp-results.txt to view the results:
less -S blastp-results.txt
The -S option of the less command disables
word-wrap.
Output:
# BLASTP 2.9.0+
# Query: sp|P0DTC2|SPIKE_SARS2 Spike glycoprotein OS=Severe acute respiratory syndrome coronavirus 2 OX=2697049 GN=S PE=1 SV=1
# Database: /home/user/databases/swissprot
# Fields: subject acc., subject title, query length, subject length, % identity
# 88 hits found
P0DTC2 Spike glycoprotein OS=Severe acute respiratory syndrome coronavirus 2 OX=2697049 GN=S PE=1 SV=1 1273 1273 100.000
P59594 Spike glycoprotein OS=Severe acute respiratory syndrome coronavirus OX=694009 GN=S PE=1 SV=1 1273 1255 76.038
Q3LZX1 Spike glycoprotein OS=Bat coronavirus HKU3 OX=442736 GN=S PE=3 SV=1 1273 1242 76.041
Q3I5J5 Spike glycoprotein OS=Bat coronavirus Rp3/2004 OX=349344 GN=S PE=1 SV=1 1273 1241 75.334
Q0Q475 Spike glycoprotein OS=Bat coronavirus 279/2005 OX=389167 GN=S PE=3 SV=1 1273 1241 74.745
...
Comments